What is the key to ensuring patient safety and global traceability for medical device product data across all manufacturers, regulatory authorities and healthcare systems? Unique Device Identification, or UDI. UDI implementation and submission is vital for manufacturers placing medical device products on the market. Let's start with fully understanding the importance of UDI requirements, challenges, and opportunities.
What is a Unique Device Identifier (UDI)?
UDI is a regulatory requirement first enacted by the US Food and Drug Administration (FDA) to serve as a system for universal tracking and labeling requirements for medical devices sold within regions around the world. Over time, the concept has expanded to other global health authorities including the European Union (EUDAMED), China NMPA, South Korea MFDS and others.
The UDI is portrayed as an alphanumeric or numeric code applied in a uniform marking system, specific to one medical device model and version only. The globally unique UDI code consists of two parts generated by the owner per an approved Issuing Agency standard (GS1, HIBCC, ICCBBA, IFA (Germany-Europe)): the Device Identifier (DI) and Production Identifier (PI).
- The DI is a fixed and mandatory portion identifying the labeler and specific model/version of a device and comes from an Issuing Agency.
- The PI is a conditional variable portion of a UDI identifying one or more of the following when included on the label of a device:
- Lot or batch number within which a device was manufactured
- Serial number of a specific device
- Expiration date of a specific device
- Date in which the device was manufactured
- Distinct identification code required by §1271.290(c) for a human cell, tissue, or cellular and tissue-based product (HCT/P) regulated as a device
These two identifiers added together create the UDI. Because the PI is conditional, it sometimes may not be included in the UDI at all. A breakdown of the UDI components is featured below.
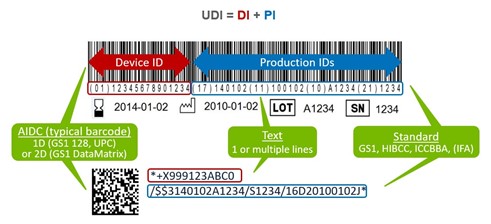
Why is a UDI required? What is its purpose?
The main regulatory goal of the UDI system is to improve patient safety. The use of a fully standardized system facilitates traceability, benefitting stakeholders in the medical device sector, including manufacturers, regulatory authorities, healthcare professionals and patients.
Benefits of a UDI System
Some of the benefits of the UDI system include:
- Increasingly more accurate reporting, reviewing and analyzing of adverse event reports to efficiently identify, address and correct malfunctioning medical devices
- Reducing medical errors by enabling health care professionals and other stakeholders to promptly identify a device and its comprehensive information and characteristics
- Enhancing analysis of devices by providing a cohesive and clear approach to document medical device use in electronic health records, clinical information systems, claim data sources and registries. Additionally, a post-market surveillance system can be utilized for premarket approval of both new and currently marketed devices
- Providing a standardized identifier to allow stakeholders (including manufacturers, distributors and healthcare facilities) to effectively manage recalls, no matter how many times the device is repackaged or re-labeled
- Creating a foundation for a globally secure distribution chain for identifying and addressing counterfeit products
- A medical device identification system recognized around the world
What are the UDI requirements?
The actual product manufacturer is responsible for creating the identifier based on an approved standard. They must provide the UDI on the label and package of a device. The UDI information is commonly presented in a linear barcode with the full UDI Device ID presented first, followed by the Production ID. Another option is a two-dimensional matrix.
It’s a requirement that the text encoded in the barcode is presented in human readable form, seen in a single line (or on multiple lines). The UDI must be printed on all single-use device labels and packaging in easy-to-read plain text and machine-readable text for automatic identification capture (AIDC) technology.
If a device is intended to be used more than once, and intended to be reprocessed before each use, it must also be directly marked with a UDI, allowing accurate identification even when the device is no longer accompanied by its label or package.
Is UDI the same as GTIN?
Global Trade Item Number (GTIN) is a globally unique identification key maintained by the GS1 organization and has applications widely used across various healthcare industries. The GTIN is one of three standards permissible by the US FDA to be used as the Device Identifier portion of the UDI. Manufacturers may use GTIN as their primary DI when exchanging product data through the Global Data Synchronization Network (GDSN). Although, merely using GTIN alone does not mean your data is in the GDSN. Product data must be placed into a GSDN-certified data pool.
UDI and Global Unique Device Identification Database
The Global Unique Device Identification Database (GUDID) is a database administered by the US FDA. Only the Device Identifier portion of the UDI is submitted and stored in the GUDID. The FDA guidance states GUDID adheres to 21 CFR 830.310, requiring electronic records to be maintained or submitted to the FDA.
How is the UDI System enforced?
Various health authorities across the globe require UDI to be reported to their database, depending on medical device class and timeline. Reed Tech has created a high-level summary of global UDI requirements per region, which currently includes these active UDI channels; US FDA, EU EUDAMED (voluntary for UDI/device module), China NMPA, South Korea MFDS, and others as they emerge.
UDI compliance for medical devices
These UDI system implementations are per published regulations, authorized by the regional health authority, with differing compliance requirements and timelines. Some of the health authority UDI requirements are listed below.
US FDA UDI Requirements – Class III, I/LS/LS and Class II devices have already been enforced. For specific scenarios for Class I devices, a UDI product data submission is due to the US FDA database, GUDID, by September 24, 2022. There are 57 UDI data attributes that must be reported to the GUDID.
European Union UDI Requirements – There are 109 data attributes required for reporting to the EU database EUDAMED, including the unique basic UDI or BUDI-DI group concept. The UDI/Device Registration and Certificate Module is currently active for voluntary use. The other modules (Clinical Investigation, Vigilance and Post-Market Surveillance and Market Surveillance) are in the plan for development. EUDAMED will provide a fully functional notice in mid-2023 with mandatory registration occurring at the end of 2023 to 2025.
China National Medical Products Administration (NMPA) UDI Requirements – This requirement was issued on August 23, 2019 with UDI rules consistent with the International Medical Device Regulators Forum (IMDRF) guidance, however no full schedule has been published. A UDI pilot, which occurred on January 1, 2021, established Class III devices (high-risk implants and instruments with 69 categories and 51 data attributes) will be in Batch 1 devices requiring UDI data label. Remaining Class III devices, not covered in Batch 1, are considered Batch 2 and will go into effect on June 22, 2022. Official notice of the UDI compliance dates for Class II and Class I devices has not yet been provided by NMPA.
South Korea Ministry of Food and Drug Safety (MFDS) UDI Requirements – Class IV, Class III and Class II UDI data is required to be on labels and reported to the Integrated Medical Device Information System (IMDIS). Class I medical device UDI data must be reported by July 1, 2022. MFDS is still accepting Class IV, III and II medical device submissions to the IMDIS database. There are 40 data attributes that must be reported to IMDIS with UDI applied to labels by class and must be submitted through a Supply Report (Track & Trace) of approximately 10 distribution metrics each month.
Challenges with enforcing the UDI system
As health authorities create and enforce differing data attributes at various timelines for UDI mandates, these variances increase complexity for manufacturers required to submit data to each jurisdiction in order to meet their individual timelines. Differing data attributes and mandate timelines can be a barrier to standards-based UDI adoption for healthcare systems and other downstream users, according to the Regulatory Affairs Professional Society (RAPS). Overall, the global UDI system sustainability depends on accurate data quality and submission reporting across all health authorities.
Stay compliant and save resources with UDI requirements by engaging with a UDI Specialist
It’s no surprise that a substantial effort is required for manufacturers to keep up to date with various global health authority regulations, timelines and complex nuances for UDI alone. As certain mandates approach, including the US FDA Class I submission requirement, you may find that enlisting the help of a UDI expert can save you time, money and bandwidth.

About LexisNexis Reed Tech
Reed Tech solutions were built to meet the product data management needs of medical device manufacturers and integrate with PTC’s Windchill PLM medical device solution.
Reed Tech serves almost half of the world’s top thirty medical device manufacturers, ranked by revenue. With experience ranging from start-up manufacturers to mid-size and industry-leading manufacturers, Reed Tech understands the challenges and serves the call for that ‘last mile of support’ for compliance. They offer solutions that help facilitate the research, collection, analysis, transformation, and submission of regulatory data to Health Authorities (US FDA, EU MDR UDI, China NMPA and others.)
