Gary Saner ist ein Experte für die eindeutige Identifizierung von Medizinprodukten und andere strukturierte Inhalte, die an Aufsichtsbehörden und kommerzielle Organisationen gemeldet werden. Er verfügt über mehr als 30 Jahre Erfahrung in den Bereichen Softwareentwicklung, Prozessmanagement und Datenverwaltung, wobei er sich in den letzten 15 Jahren auf die Life Sciences-Branche konzentriert hat. Er ist Co-Vorsitzender des Structured Product Labeling Technical Team der Branche und Mitglied des Beirats der Medical Devices Group.
Was ist der Schlüssel zur Gewährleistung der Patientensicherheit und der globalen Rückverfolgbarkeit der Produktdaten von Medizinprodukten über alle Hersteller, Aufsichtsbehörden und Gesundheitssysteme hinweg? Die einmalige Produktkennung (Unique Device Identification, UDI). Die Implementierung und Einreichung von UDI ist für Hersteller, die Medizinprodukte auf den Markt bringen, unerlässlich. Lassen Sie uns damit beginnen, die Bedeutung der UDI-Anforderungen, Herausforderungen und Möglichkeiten zu verstehen.
Was ist die einmalige Produktkennung (UDI)?
UDI ist eine gesetzliche Vorschrift, die zunächst von der US-amerikanischen Food and Drug Administration (FDA) eingeführt wurde, um ein System für die universelle Rückverfolgung und Kennzeichnung von Medizinprodukten zu schaffen, die in verschiedenen Regionen der Welt verkauft werden. Im Laufe der Zeit wurde das Konzept auf andere globale Gesundheitsbehörden ausgeweitet, darunter die Europäische Union (EUDAMED), China NMPA, Südkorea MFDS und andere.
Die UDI wird als alphanumerischer oder numerischer Code in einem einheitlichen Kennzeichnungssystem dargestellt, das nur für ein bestimmtes Modell und eine bestimmte Version eines Medizinprodukts gilt. Der weltweit eindeutige UDI-Code besteht aus zwei Teilen, die vom Eigentümer nach einem genehmigten Standard der Ausgabestelle (GS1, HIBCC, ICCBBA, IFA (Deutschland-Europa)) generiert werden: dem Device Identifier (DI) und dem Production Identifier (PI).
- Die DI ist ein fester und obligatorischer Teil, der den Etikettierer und das spezifische Modell/die Version eines Geräts identifiziert und von einer ausstellenden Agentur stammt.
- Der PI ist ein bedingter variabler Teil einer UDI, der eine oder mehrere der folgenden Angaben enthält, wenn er auf dem Etikett eines Produkts angegeben ist:
- Los- oder Chargennummer, unter der ein Produkt hergestellt wurde
- Seriennummer eines bestimmten Geräts
- Verfallsdatum eines bestimmten Geräts
- Datum, an dem das Gerät hergestellt wurde
- Eindeutiger Identifizierungscode gemäß §1271.290(c) für ein menschliches Zell-, Gewebe- oder zell- und gewebebasiertes Produkt (HCT/P), das als Produkt geregelt ist
Diese beiden Identifikatoren ergeben zusammen die UDI. Da der PI an Bedingungen geknüpft ist, kann es vorkommen, dass er gar nicht in die UDI aufgenommen wird. Nachstehend finden Sie eine Aufschlüsselung der UDI-Komponenten.
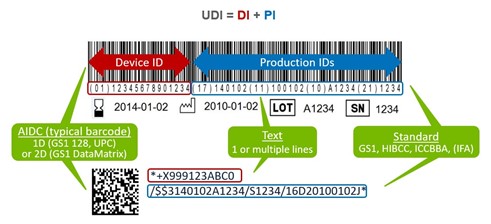
Warum ist eine UDI erforderlich? Was ist ihr Zweck?
Das wichtigste regulatorische Ziel des UDI-Systems ist die Verbesserung der Patientensicherheit. Die Verwendung eines vollständig standardisierten Systems erleichtert die Rückverfolgbarkeit, wovon alle Beteiligten im Medizinproduktesektor, einschließlich Hersteller, Regulierungsbehörden, Beschäftigte im Gesundheitswesen und Patienten, profitieren.
Vorteile eines UDI-Systems
Zu den Vorteilen des UDI-Systems gehören:
- Zunehmend genauere Erfassung, Überprüfung und Analyse von Berichten über unerwünschte Ereignisse, um Fehlfunktionen von Medizinprodukten effizient zu erkennen, zu beheben und zu korrigieren
- Verringerung medizinischer Fehler, indem medizinisches Fachpersonal und andere Beteiligte in die Lage versetzt werden, ein Produkt und seine umfassenden Informationen und Merkmale sofort zu erkennen
- Verbesserung der Analyse von Produkten durch Bereitstellung eines kohärenten und klaren Ansatzes zur Dokumentation der Verwendung von Medizinprodukten in elektronischen Gesundheitsakten, klinischen Informationssystemen, Anspruchsdatenquellen und Registern. Außerdem kann ein System zur Überwachung nach dem Inverkehrbringen für die Zulassung neuer und bereits auf dem Markt befindlicher Produkte genutzt werden.
- Bereitstellung einer standardisierten Kennung, die es den Beteiligten (einschließlich Herstellern, Händlern und Gesundheitseinrichtungen) ermöglicht, Rückrufe effektiv zu verwalten, unabhängig davon, wie oft das Produkt neu verpackt oder neu etikettiert wird.
- Schaffung einer Grundlage für eine weltweit sichere Vertriebskette zur Erkennung und Bekämpfung von Produktfälschungen.
- Ein weltweit anerkanntes System zur Identifizierung von Medizinprodukten.
Was sind die UDI-Anforderungen?
Der eigentliche Produkthersteller ist für die Erstellung der Kennung auf der Grundlage eines genehmigten Standards verantwortlich. Er muss die UDI auf dem Etikett und der Verpackung eines Produkts angeben. Die UDI-Informationen werden in der Regel in einem linearen Barcode dargestellt, wobei die vollständige UDI-Geräte-ID an erster Stelle steht, gefolgt von der Produktions-ID. Eine andere Möglichkeit ist eine zweidimensionale Matrix.
Der im Barcode kodierte Text muss in einer einzigen Zeile (oder in mehreren Zeilen) in für den Menschen lesbarer Form dargestellt sein. Die UDI muss auf allen Etiketten und Verpackungen von Einwegprodukten in leicht lesbarem Klartext und maschinenlesbarem Text für die automatische Identifikationserfassung (AIDC) aufgedruckt sein.
Ist ein Produkt zur mehrmaligen Verwendung bestimmt und soll es vor jeder Verwendung wieder aufbereitet werden, muss es auch direkt mit einer UDI gekennzeichnet werden, so dass eine genaue Identifizierung möglich ist, auch wenn das Produkt nicht mehr von seinem Etikett oder seiner Verpackung begleitet wird.
Ist UDI dasselbe wie die GTIN?
Die Global Trade Item Number (GTIN) ist ein weltweit eindeutiger Identifikationsschlüssel, der von der GS1-Organisation verwaltet wird und in verschiedenen Branchen des Gesundheitswesens breite Anwendung findet. Die GTIN ist einer von drei Standards, die von der US-amerikanischen FDA für die Verwendung als Device Identifier-Teil der UDI zugelassen sind. Hersteller können die GTIN als primäre DI beim Austausch von Produktdaten über das Global Data Synchronization Network (GDSN) verwenden. Die Verwendung der GTIN allein bedeutet jedoch nicht, dass Ihre Daten im GDSN enthalten sind. Die Produktdaten müssen in einen GSDN-zertifizierten Datenpool eingestellt werden.
UDI und die globale Datenbank zur eindeutigen Geräteidentifizierung
Die Global Unique Device Identification Database (GUDID) ist eine Datenbank, die von der US-FDA verwaltet wird. In der GUDID wird nur der Teil der Gerätekennung (Device Identifier) der UDI übermittelt und gespeichert. Der FDA-Leitfaden besagt, dass GUDID der Vorschrift 21 CFR 830.310 entspricht, nach der elektronische Aufzeichnungen geführt oder an die FDA übermittelt werden müssen.
Wie wird das UDI-System durchgesetzt?
Verschiedene Gesundheitsbehörden auf der ganzen Welt verlangen die Meldung von UDI an ihre Datenbank, abhängig von der Klasse der Medizinprodukte und dem Zeitplan. Reed Tech hat eine Übersicht über die weltweiten UDI-Anforderungen pro Region erstellt, die derzeit die folgenden aktiven UDI-Kanäle umfasst: US FDA, EU EUDAMED (freiwillig für UDI/Gerätemodul), China NMPA, Südkorea MFDS und weitere, die noch hinzukommen.
UDI Compliance für Medizinprodukte
Diese UDI-Systeme werden auf der Grundlage veröffentlichter und von der regionalen Gesundheitsbehörde genehmigter Vorschriften mit unterschiedlichen Anforderungen und Fristen für die Einhaltung der Vorschriften eingeführt. Einige der UDI-Anforderungen der Gesundheitsbehörde sind unten aufgeführt.
Die UDI-Anforderungen der US-FDA für Produkte der Klassen III, I/LS/LS und II sind bereits in Kraft getreten. Für bestimmte Szenarien für Produkte der Klasse I müssen bis zum 24. September 2022 UDI-Produktdaten an die US-FDA-Datenbank GUDID übermittelt werden. Es gibt 57 UDI-Datenattribute, die an die GUDID gemeldet werden müssen.
UDI-Anforderungen der Europäischen Union – Es gibt 109 Datenattribute, die für die Meldung an die EU-Datenbank EUDAMED erforderlich sind, einschließlich des eindeutigen UDI-Basiskonzepts oder BUDI-DI-Gruppenkonzepts. Das Modul UDI/Device Registration and Certificate ist derzeit für die freiwillige Nutzung aktiv. Die anderen Module (Klinische Prüfung, Vigilanz und Überwachung nach dem Inverkehrbringen und Marktüberwachung) sind in der Entwicklung begriffen. EUDAMED wird Mitte 2023 eine voll funktionsfähige Bekanntmachung bereitstellen, wobei die obligatorische Registrierung Ende 2023 bis 2025 erfolgen wird.
China National Medical Products Administration (NMPA) UDI-Anforderungen – Diese Anforderung wurde am 23. August 2019 mit UDI-Regeln veröffentlicht, die mit den Leitlinien des International Medical Device Regulators Forum (IMDRF) übereinstimmen, jedoch wurde kein vollständiger Zeitplan veröffentlicht. Im Rahmen eines UDI-Pilotprojekts, das am 1. Januar 2021 stattfand, wurde festgelegt, dass Produkte der Klasse III (Hochrisikoimplantate und -instrumente mit 69 Kategorien und 51 Datenattributen) zu den Produkten der Charge 1 gehören, für die eine UDI-Datenkennzeichnung erforderlich ist. Die übrigen Produkte der Klasse III, die nicht unter Charge 1 fallen, gelten als Produkte der Charge 2 und werden am 22. Juni 2022 in Kraft treten. Die NMPA hat die UDI-Konformitätstermine für Produkte der Klassen II und I noch nicht offiziell bekannt gegeben.
Südkoreas Ministerium für Lebensmittel- und Arzneimittelsicherheit (MFDS) UDI-Anforderungen – UDI-Daten der Klassen IV, III und II müssen auf Etiketten angegeben und an das Integrierte Informationssystem für Medizinprodukte (IMDIS) gemeldet werden. UDI-Daten für Medizinprodukte der Klasse I müssen bis zum 1. Juli 2022 gemeldet werden. Das MFDS nimmt weiterhin Meldungen von Medizinprodukten der Klassen IV, III und II an die IMDIS-Datenbank entgegen. Es gibt 40 Datenattribute, die an IMDIS gemeldet werden müssen, wobei die UDI auf die Etiketten nach Klassen angewandt wird, und die durch einen Lieferbericht (Track & Trace) mit etwa 10 Vertriebsmetriken pro Monat übermittelt werden müssen.
Herausforderungen bei der Durchsetzung des UDI-Systems
Da die Gesundheitsbehörden unterschiedliche Datenattribute zu verschiedenen Zeitpunkten für UDI-Mandate erstellen und durchsetzen, erhöhen diese Abweichungen die Komplexität für die Hersteller, die Daten an jede Gerichtsbarkeit übermitteln müssen, um ihre individuellen Zeitpläne einzuhalten. Laut der Regulatory Affairs Professional Society (RAPS) können unterschiedliche Datenattribute und Mandatsfristen ein Hindernis für die standardbasierte UDI-Einführung für Gesundheitssysteme und andere nachgeschaltete Anwender darstellen. Insgesamt hängt die Nachhaltigkeit des globalen UDI-Systems von der genauen Datenqualität und der Einreichung von Berichten durch alle Gesundheitsbehörden ab.
Halten Sie die UDI-Anforderungen ein und sparen Sie Ressourcen, indem Sie sich an einen UDI-Spezialisten wenden.
Es ist nicht verwunderlich, dass es für Hersteller einen erheblichen Aufwand bedeutet, sich mit den verschiedenen Vorschriften der globalen Gesundheitsbehörden, den Fristen und den komplexen Nuancen allein für UDI auf dem Laufenden zu halten. Da bestimmte Vorschriften näher rücken, darunter die US-FDA-Vorschrift zur Einreichung der Klasse I, werden Sie feststellen, dass die Inanspruchnahme der Hilfe eines UDI-Experten Ihnen Zeit, Geld und Ressourcen sparen kann.
--
Über LexisNexis Reed Tech
Die Lösungen von Reed Tech wurden speziell für die Anforderungen von Herstellern medizinischer Geräte an das Produktdatenmanagement entwickelt und lassen sich in die Windchill PLM-Lösung für medizinische Geräte von PTC integrieren.
Reed Tech beliefert fast die Hälfte der dreißig weltweit führenden Hersteller von Medizinprodukten (nach Umsatz). Mit seiner Erfahrung, die von Start-up-Herstellern bis hin zu mittelgroßen und branchenführenden Herstellern reicht, kennt Reed Tech die Herausforderungen und erfüllt die Forderung nach der "last mile of support" für die Einhaltung der Vorschriften. Das Unternehmen bietet Lösungen an, die die Recherche, Sammlung, Analyse, Umwandlung und Einreichung von Zulassungsdaten bei Gesundheitsbehörden (US FDA, EU MDR UDI, China NMPA und andere) erleichtern.

Gary Saner ist ein Experte für die eindeutige Identifizierung von Medizinprodukten und andere strukturierte Inhalte, die an Aufsichtsbehörden und kommerzielle Organisationen gemeldet werden. Er verfügt über mehr als 30 Jahre Erfahrung in den Bereichen Softwareentwicklung, Prozessmanagement und Datenverwaltung, wobei er sich in den letzten 15 Jahren auf die Life Sciences-Branche konzentriert hat. Er ist Co-Vorsitzender des Structured Product Labeling Technical Team der Branche und Mitglied des Beirats der Medical Devices Group.